| Chapter one: Introduction | Page no. | |
| Abstract | 4 | |
| 1 | Introduction | 6 |
| 1.1 | Preview | 7 |
| 1.1.2 | History | 7 |
| 1.1.3 | Medical Use | 7 |
| 1.1.4 | Classification | 7-8 |
| 1.1.5 | Sign and Symptoms | 8 |
| 1.1.6 | Side effects | 8 |
| 1.1.7 | Management | 8 |
| 1.1.8 | Prevention | 8 |
| 1.2 | Chemistry of drug | 8 |
| 1.2.1 | Chemical Name | 8 |
| 1.2.2 | Molecular Formula | 9 |
| 1.2.3 | Molecular Weight | 9 |
| 1.2.4 | Structure | 9 |
| 1.2.5 | Synthesis | 9 |
| 1.3 | Physical and Chemical Properties | 10 |
| 1.3.1 | Appearance | 10 |
| 1.3.2 | Taste | 10 |
| 1.3.3 | Solubility | 10 |
| 1.3.4 | Melting Point | 10 |
| 1.3.5 | Boiling Point | 10 |
| 1.3.6 | Density | 10 |
| 1.4 | Pharmacology and Biopharmaceutics | 10 |
| 1.4.1 | Pharmacodynamics | 10 |
| 1.4.1.1 | Receptor-Binding Affinity | 10 |
| 1.4.1.2 | Receptor Selectivity | 11 |
| 1.4.1.3 | Anti-Inflammatory Effects | 12 |
| 1.4.1.4 | CNS Effects | 13 |
| 1.4.2 | Pharmacokinetics | 14 |
| 1.4.2.1 | Absorption | 14 |
| 1.4.2.2 | Distribution | 14 |
| 1.4.2.3 | Metabolism | 14 |
| 1.4.2.4 | Elimination | 14 |
| 1.5 | Indication | 15 |
| 1.6 | Side Effect | 15 |
| 1.7 | Contraindications | 16 |
| 1.8 | Warning and Precaution | 16 |
| 1.9 | Toxicology Studies | 16 |
| 1.10 | Food And Drug Interaction | 17 |
| 1.11 | Use Of Desloratadine In Special Patient Subpopulations | 17 |
| 1.12 | Poor Metabolizers | 18-19 |
| 1.13 | Pharmaceutical Technology | 19 |
| 1.13.1 | Dosage Form | 19 |
| 1.13.2 | Available Dosage | 19 |
| 1.13.3 | Dosage | 19 |
| 1.14 | Quality Parameters Tested | 19 |
| 1.14.1 | Thickness and diameter | 19 |
| 1.14.2 | Hardness | 19-20 |
| 1.14.3 | Friability Test | 21 |
| 1.14.4 | Weight Variation | 21 |
| 1.14.5 | Disintegration test | 22 |
| 1.14.6 | Dissolution | 22 |
| 1.15 | purpose of the study | 23 |
| Chapter two: Materials and methods | ||
| 2.1 | Apparatus | 24 |
| 2.2 | Weight variation | 25 |
| 2.3 | Thickness test | 26 |
| 2.4 | Diameter test | 27 |
| 2.5 | Friability test | 28 |
| 2.6 | Hardness test | 28 |
| 2.7 | Disintegration test | 29 |
| 2.8 | Disintegration Apparatus | 30 |
| 2.9 | Dissolution Test | 31 |
| 2.10 | Potency | 32 |
| Chapter three: Results and discussion | ||
| 3 | Aesthetic test | 36 |
| 3.1 | Friability | 36 |
| 3.2 | Thickness | 37 |
| 3.3 | Hardness | 38 |
| 3.4 | Diameter | 38 |
| 3.6 | conclusion | 40 |
| 3.7 | Reference | 41-44 |
| List of Tables | ||
| 1 | Antihistamines Classification | 8 |
| 2 | Apparatus used for the test | 23 |
| 3 | Concentration of drugs for standard curve | 31 |
| 4 | Results of aesthetic tests of the tablets of three different brands | 34 |
| 5 | Comparison of the thickness of 3 different brands of desloratadine. | 35 |
| List of Figures | ||
| 1.1.4 | Structure | 9 |
| 2.1 | Electronic Balance | 27 |
| 2.2 | Digital Slide Calipers | 28 |
| 2.3 | Electrolab Friabilator | 29 |
| 2.4 | Hardness Tester | 29 |
Abstract:
A series of desloratadine derivatives were stereoselectively synthesized and evaluated for H1 antihista mine activity. For the evaluation of H1 antihistamine activity, the in vitro histamine induced contraction of the guinea pig ileum assay (HC) was used. The synthesized desloratadine derivatives 7, 8 and 9 are structurally related to rupatadine and were generated by replacement of the 5-methyl-3-pyridine group of rupatadine with c-alkylidene butenolide. Their H1 antihistamine activities have shown a high dependence on the exact nature of the substituent in the lactone ring. Optimum structures 7, 8a and 8g display potent activity inhibiting histamine- induced effects.Second generation histamine H1 receptor antagonists were developed to provide efficacious treatment of allergic rhinitis (AR) and chronic idiopathic urticaria (CIU) while decreasing adverse effects associated with first generation agents. As a class, second generation antihistamines are highly selective for the H1 receptor. Some bind to it with high affinity, although there is marked heterogeneity among the various compounds. They have a limited effect on the CNS, and clinical studies have noted almost no significant drug drug interactions in the agents studied. No major cytochrome P450 inhibition has been reported with desloratadine, and the bioavailability of desloratadine is minimally affected by drugs interfering with transporter molecules. Of the second generation antihistamines, desloratadine has the greatest binding affinity for the H1 receptor. Since desloratadine is the principal metabolite of loratadine, it can be assumed that a similar safety profile would fit for desloratadine as was described for loratadine although no direct human studies have been done. In a meta analysis examining the safety of firstgeneration antihis tamines in pregnancy, 200 000 first trimester exposures failed to show increased teratogenic risk. The aim of this work was the development and validation of a dissolution methodology for desloratadine coated tablets by spectrophotometry on ultraviolet (UV). 0.1M hydrochloric acid (HCl), pH 4.5-citrate buffer and pH 6.8 phosphate buffer were tested as dissolution medium. In addition, influences of apparatus, and rotation speed were evaluated. After an UV scan spectrum from 500 to 200 nm, The parameters selected were 0.1 M HCl as dissolution medium, using paddles as apparatus at 50 rpm, with analysis at wavelength of 280 nm with sampling points at 5,10,15 and 30 minutes is specific.
Chapter 1
Introduction
1. Introduction: Desloratadine (descarboethoxyloratadine) is a selective, non-selective, second-generation tricyclic antihistamine which has a selective and peripheral antagonistic action. Desloratadin is a long acting piperidine derivatives which is chemically similar to loratadine (Etman et al.,2014). Studies in mice have shown that desloratadine is approximately four time as potent as loratadine, whereas human in vitro studies have indicated that it is up to 10 time more active(Gupta et al.,2007). Histamine is a chemical that is responsible for many signs and symptoms caused by released of histamine. Desloratadine is a international Nonproprietary Name (INN)drug and was approved by the Food and Drug Administration(FDA) on December 21,2001(Buck,2011). Desloratadine is well absorbed from the gut and reaches highest blood plasma concentrations after about three hours. In the bloodstream, 83 to 87% of the substance are bound to plasma proteins(2022-01-21).Desloratadine is metabolized to 3-hydroxydesloratadine in a three-step sequence in normal metabolizers. First, n-glucuronidation of desloratadine by UGT2B10; then, 3-hydroxylation of desloratadine N-glucuronide by CYP2C8; and finally, a non-enzymatic deconjugation of 3-hydroxydesloratadine N-glucuronide.[13] Both desloratadine and 3-hydroxydesloratadine are eliminated via urine and feces with a half-life of 27 hours in normal metabolizers (2022-01-21). It exhibits only peripheral activity since it does not readily cross the blood-brain barrier; hence, it does not normally cause drowsiness because it does not readily enter the central nervous system. Desloratadine does not have a strong effect on a number of tested enzymes in the cytochrome P450 system. It was found to weakly inhibit CYP2B6, CYP2D6, and CYP3A4/CYP3A5, and not to inhibit CYP1A2, CYP2C8, CYP2C9, or CYP2C19.
Desloratadine was found to be a potent and relatively selective inhibitor of UGT2B10, a weak to moderate inhibitor of UGT2B17, UGT1A10, and UGT2B4, and not to inhibit UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, UGT2B15, UGT1A7, and UGT1A8.2% of Caucasian people and 18% of people from African descent are desloratadine poor metabolizers. In these people, the drug reaches threefold highest plasma concentrations six to seven hours after intake, and has a half-life of about 89 hours. However, the safety profile for these subjects is not worse than for extensive(normal)metabolizer.
1.1 preview
It was a popular view a generation ago that a non-sedating H1 receptor antagonist is unobtainable. There was a lack of validated methods to predict the sedative liability of a new antihistamine. Terfenadine served as a clinical breakthrough, which proved to be such an agent in 1978. This initiated researchers at Schering-Plough to carry out drug analog research, whose lead molecules were terfenadine and azatadine. A selected battery of central nervous system(CNS)tests in guinea pigs and mice using terfenadine and azatadine as reference drugs helped them to screen analogs. The carbamate analog of azatadine removed its CNS activity while retaining much of its antihistamine potency. Further optimizing the carbamate azatadine afforded the 8-chloro derivatives loratadine, with a longer duration of action. Its active metabolite, desloratadine was lunched in 2001.(Fischer and Gere,2006).
1.1.2 History: Desloratadine (trade names Clarinex and Aerius) is a tricyclic H1 inverse agonist that is used to treat allergies. It is an active metabolite of loratadine.It was patented in 1984 and came into medical use in 2001(Fischer, Jnos;et al.,2006).
1.1.3 Medical Use: Desloratadine is used to treat allergic rhinitis, nasal congestion and chronic idiopathic urticaria (hives)(2003). It is the major metabolite of loratadine and the two drugs are similar in safety and effectiveness(2003). Desloratadine is available in many dosage forms and under many trade names worldwide(Drugs.com). An emerging indication for desloratadine is in the treatment of acne, as an inexpensive adjuvant to isotretinoin and possibly as maintenance therapy or monotherapy (Lee HE,et al 2014).
1.1.4 Classification: H1 blockers have 2 generations–Generation 1(like diphenhydramine, hydroxyzine) are older, and they also depress the CNS and produce sedation and disorientation. Generation 2 (like levocetrizine, fexofenadine, loratadine, desloratadine) are newer and less sedating, so they are preferred. The drugs which block H2 receptors mainly decrease the gastric acid secretion and are usually identified by the name “H2 blockers” such as ranitidine, famotidine etc (2019/10/06).
1.Antihistamines Classification
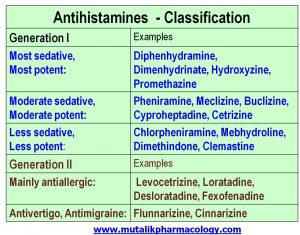
1.1.5 Sign and Symptoms: rash,itching,hives,difficulty breathing or swallowing,swelling of the face, throat, tongue, lips, eyes, hands, feet, ankles, or lower legs.
1.1.6 Side effects:headache,nausea,diarrhea,dizziness,sore throat,dry mouth,muscle pain,extreme, tiredness,sleepiness,painful menstruation.
1.1.7 Management: Recommended doses of desloratadine are:
- For adults and for children aged over 12 years: 5 mg taken once a day.
- For children aged 6-12 years: 2.5 mg taken once a day. This is 5 ml of the liquid medicine.
- For children aged 1-6 years: 1.25 mg taken once a day. This is 2.5 ml of the liquid medicine.
1.1.8 Prevention: Most people only need to take an antihistamine for a short while when they have symptoms. You should stop taking desloratadine once your symptoms have eased.
Although desloratadine is classed as a non-drowsy antihistamine, it can still cause drowsiness in a few people. If this happens to you, do not drive and do not use tools or machines.Desloratadine is called a non-drowsy antihistamine; however, it can still cause drowsiness in a few people.
Desloratadine is an antihistamine. It is used to relieve the symptoms of hay fever and hives of the skin. Antihistamines work by preventing the effects of a substance called histamine, which is produced by the body. Histamine can cause itching, sneezing, runny nose, and watery eyes.
- Chemistry of Drug
- Chemical Name: 8-chloro-11-piperidin-4-ylidene-5,6-dihydrobenzo[1,2]cyclohepta[2,4- b]pyridine.
- Molecular Formula: C₁₉H₁₉CIN₂
- Molecular Weight: 310.82 g/mol.
- Structure
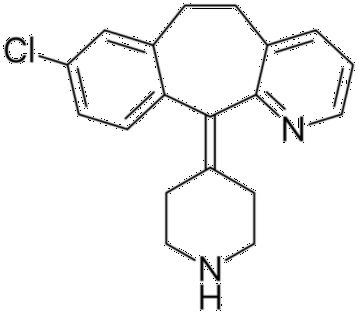
- Synthesis
A solution of loratadine(38.2 g, 0.1 mol) and 15 equiv KOH (84 g, 1.5 mol) in 80% CH3CH2OH (334 ml) was heated under reflux for 10 h and the reaction was monitored by TLC to completion.
Ethyl alcohol was evaporated under reduced pressure, and then ex- tracted with ethyl acetate (3100 ml), and the combined extracts were washed with saturated NaHCO3 (240 ml) and brine (2100 ml) and dried over anhydrous Na2SO4, the solution was evaporated under reduced pressure to give a crude desloratadine, which was refined by recrystallization in ethyl acetate to give 27 g white crystal. Yield: 88%( Liu et al.,2010).
- Physical and Chemical properties
- Appearance: Beige Solid
- Taste : Bitter Taste.
- Solubility : >10mg/mL is soluble in DMSO , Freely soluble in methanol.
- Melting Point: 150-151°C.
- Boiling Point: 467.9 °C at 760 mmHg.
- Density: 1.221 g/cm3.
- Vapour Pressure: 6.25E-09 mmHg at 25 °C.
- Pharmacology and Biopharmaceutics
- Pharmacodynamics
- Receptor-Binding Affinity
Desloratadine has a higher H1 receptor affinity than other second-generation antihistamines at nearly 200 times that of fexofenadine, >50 times that of loratadine and cetirizine, and three times that of levocetirizine(Anthes et al.,2002;). In an in vitro study of inverse agonism, desloratadine appeared to be more potent than fexofenadine and cetirizine, a result explainable by the high correlation between H1 receptor affinity and inverse agonist activity(Bakker et al.,2000).
1.3.1.2. Receptor Selectivity
Desloratadine has >60 times higher affinity for H1 receptors than it has for H2 receptors(Kreutner et al.,2000). The human H1 receptor has 45% sequence homology with the muscarinic receptor; thus, there is the potential for interaction between H1 receptor antagonists
and muscarinic receptors. This interaction results in anticholinergic effects such as dry mouth
and urinary retention, as noted with non-H1 receptor-selective first- generation H1 receptor antagonists(de Esch et al.,2005). The affinity of desloratadine for muscarinic receptors is about 50- to 100-fold lower than its affinity for H1 receptors(Liu et al.,2005). Peak plasma concentrations of desloratadine at recommended doses for antihistaminic activity are 10-fold lower than the concentrations at which in vitro functional antimuscarinic activities are observed. Desloratadine does not induce any clinically relevant antimuscarinic effects at therapeutic doses(Kerutner et al., 2000;).
- Anti-Inflammatory Effects
Mediators other than histamine are involved in both the early and late phases of the allergic response. In the early phase response (Bousquet et al.,2001), inflammatory mediators, such as histamine, leukotrienes, bradykinin, cytokines and platelet-activating factor, are released from inflammatory cells. These mediators stimulate in creased vascular permeability and vasodilatation within the nasal mucosa, which leads to nasal oedema and congestion. They also contribute to sneezing, pruritus, rhinorrhoea and ocular itching, redness and tearing. The late phase allergic response generally begins 2–4 hours after the early phase response and can last for up to 24 hours. During this phase, inflammatory cytokines produced in the early phase response stimulate adhesion of circulating leukocytes and infiltration of tissues by eosinophils, neutrophils and basophils. The activated infiltrating inflammatory cells release mediators that prolong and enhance the allergic cascade. Pharmacological studies suggest that H1 receptor antagonists are able to exert anti-inflammatory effects(Golightly, Greos,2005). One study demon strated that desloratadine (1–10 mol/L) inhibits both IgE-medi ated and non-IgE-mediated generation of IL-4 and IL-13 by human basophils in vitro(Schroeder et al.,2001). Desloratadine exert anti- inflammatory effects by inhibiting the release of preformed and de novo synthesized mediators from human FcɛRI+cells(Genovese et al.,1997).The release of proinflammatory mediators from eosinophils, which are typically linked with late phase allergic reactions, is inhibited by desloratadine. At nanomolar concentrations (10–9 to 10–5 mol/L), deslorata dine has been shown to inhibit endothelial expression of P-selectin (a surface molecule involved in the adhesion of neutrophils and eosinophils to endothelial cells) and to reduce the expression of IL-6 and IL-8 in response to the histamine challenge. When preincubated with human mast and basophilic cells, desloratadine (10–11 to 10–5 mol/L) inhibited IL-6 release by 40% and IL-8 release by 50%.
Desloratadine also significantly reduced secretion of tumour necrosis factor from human leukaemic mast cells and basophils(Lippert et al.,2000).
- CNS Effects
Low blood brain barrier penetration explains the relative lack of CNS effects associated with second-generation H1 receptor antagonists, unlike their first-generation counterparts(Tashiro et al.,2005). Recent developments have shown that the CNS concentrations of drugs are controlled by the P-glycoprotein (P-gp) transporter system, as well as lipophilicity and ionization.( Pagliara et al.,1999). The newer antihistamines are usually non sedating at the recommended doses, perhaps as a result of active efflux of the drugs from the brain by P-gp (seen in P-gp transfected cells and P-gp knockout mice) (Chen et al.,2004). Because P-gp is just one of the many efflux transporters that may control the brain distribution of H1 receptor antagonists, a comparison of antihistamines based on efflux transporters would be incomplete. Also, both influx and efflux transporters are involved in the distribution of H1 receptor antagonists in the brain and should be investigated in conjunction with one another. The complexity of these mechanisms emphasizes the need for an accurate clinical evaluation of the sedative properties of antihistamines. A single center, randomized, double-blind, placebo-controlled, crossover investigation of fexofenadine hydrochloride 180mg alone and with alcohol, with hydroxyzine hydrochloride 50mg as a positive internal control, on aspects of cognitive and psychomotor function related to driving a car, and is also nonsedating, even at nine times its standard dose(Kerutner et al.,2000). In a double-blind study comparing desloratadine, levocetirizine and placebo, the incidence of somnolence with these agents was 0.7%, 2.1% and 1.1%, respectively ( Day, Briscoe, Rafeiro et al.,2004). A prescription-event monitoring study, described as an investigation of the safety of newly marketed drugs conducted under ‘primary care conditions’, reviewed adverse-event reporting among subjects receiving desloratadine(Layton,Wilton,Boshier et al.,2006). A focus of this study was to determine whether desloratadine and levocetirizine differed in their potential to cause drowsiness and sedation. The incidence of drowsiness/somnolence for desloratadine and levocetirizine was very low for both drugs: nine cases (0.1%) and 46 cases (0.4%), respectively, with 50% of cases occurring by days 7 and 14, respectively. However, the time-to-event estimates differed (log rank test, p < 0.0001).
- Pharmacokinetics
- Absorption
The pharmacokinetic feature classically considered for the evaluation of the onset of action of a drug is the time it takes to reach the peak plasma concentration (tmax). This time for desloratadine (about 3 hours) .The tmax depends on the mechanisms of the absorption phase and the level of intestinal and liver first-pass metabolisms. The onset of action of antihistaminic activity can also be evaluated through the inhibitory effects on histamine-induced skin wheal- and-flare responses. The time to reach maximal inhibition of wheals was 4 hours for desloratadine. Allergen-challenge chamber studies are often used to determine the onset of action(Aerius 2006). Absorption tmax~3 (Aerius 2006).
- Distribution
Desloratadine has a high volume of distribution (Vd) which is ~49 L/kg (Molimard et al., 2004;)
- Metabolism
Loratadine is metabolized to the active metabolite desloratadine, this metabolite are the actual drug is then metabolized to 3-hydroxydesloratadine in the liver and undergoes further metabolism by glucuronidation. The enzyme pathway(Affrime et al.,2002), involved in the liver metabolism of desloratadine to 3-hydroxydesloratadine has not yet been reported. One study demonstrated that the in vitro formation of 3-hydroxydesloratadineglucuronide from 3- hydroxydesloratadine was mediated via uridine diphosphate glucuronosyltransferase (UGT) 1A1, UGT1A3 and UGT2B15 in the human liver. Desloratadine undergoes less first-pass metabolism than loratadine.
- Elimination
Elimination occurs via both the renal and faecal pathways(Affrime et al.,2002). Urinary excretion (41% of radioactive dose) , faecal excretion (47% of radioactive dose) (US FDA ). The terminal elimination half-life (t1/2) can be used as an alternative measure of duration of action. The t1/2 of desloratadine is approximately 27 hours, compared with 11–15 hours for fexofenadine and 8 hours for levocetirizine.(Aerius,2006).
- Indication
Desloratadine has demonstrated clinical efficacy in augmented reality chronic idiopathic urticaria (CIU), and seasonal asthma. It has several advantages over other H1 antagonists in that it has proven decongestant activity, a sparing effect on the use of bronchodilators (b2-agonists) and a low potential for drug interactions(Liu et al.,2010).
- Side Effect
Fast, pounding, or uneven heartbeat;fever, flu symptoms;seizure (convulsions); or jaundice (yellowing of the skin or eyes). Less serious side effects may include:dry mouth, sore throat, cough;muscle pain ,drowsiness, tired feeling;nausea, diarrhea; orheadache.
More common:
Headache Less common:
Dizziness,dry mouth,dysmenorrhea, such as, difficult or painful menstruation,dyspepsia, such as, acid or sour stomach, belching, heartburn, indigestion, stomach discomfort , upset or pain,fatigue, such as, unusual tiredness or weakness,myalgia, such as, joint pain, swollen joints, muscle aching or cramping, muscle pains or stiffness, difficulty in moving
pharyngitis, such as, body aches or pain, congestion, cough, dryness or soreness of throat, fever, hoarseness, runny nose, tender swollen glands in neck, trouble in swallowing, voice changes.somnolence, such as, sleepiness or unusual drowsiness.
Rare:
Anaphylaxis, such as, cough, difficulty swallowing, dizziness, fast heartbeat, hives, itching, puffiness or swelling of eyelids or around the eyes or face or lips or tongue, shortness of breath, skin rash, tightness in chest, unusual tiredness or weakness, wheezing
dyspnea, such as, shortness of breath, difficult or labored breathing, tightness in chest, wheezing,pruritus, such as, itching skin,rash,tachycardia, such as, fast, pounding, or irregular heartbeat or pulse urticaria, such as, hives or welts, itching, redness of skin, skin rash.
- Contraindications
Desloratadine is contraindicated in patients who are hypersensitive to the drug.Hypersensitivity reactions including rash, pruritus, urticaria, edema, dyspnea, and anaphylaxis have been reported after administration of Desloratadine. If such a reaction occurs, therapy with Desloratadine should be stopped and alternative treatment should be considered.
- Warning and Precaution
Desloratadine is excreted into human milk. The effects in the nursing infant are unknown. The manufacturer recommends that due to the potential for serious adverse reactions in nursing infants, a decision should be made to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Desloratadine has been assigned to pregnancy category C by the FDA. Animal studies have failed to reveal evidence of teratogenicity. There are no controlled data in human pregnancy. Desloratadine is only recommended for use during pregnancy when benefit outweighs risk.
- Toxicology Studies
Desloratadine was not teratogenic in rats at doses up to 48 mg/kg/day (estimated Desloratadine and Desloratadine metabolite exposures were approximately 210 times the AUC in humans at the recommended daily oral dose) or in rabbits at doses up to 60 mg/kg/day (estimated Desloratadine exposures were approximately 230 times the AUC in humans at the recommended daily oral dose). In a separate study, an increase in pre-implantation loss and a decreased number of implantations and fetuses were noted in female rats at 24 mg/kg (estimated Desloratadine and Desloratadine metabolite exposures were approximately 120 times the AUC in humans at the recommended daily oral dose). Reduced body weight and slow righting reflex were reported in pups at doses of 9 mg/kg/day or greater (estimated Desloratadine and Desloratadine metabolite exposures were approximately 50 times or greater than the AUC in humans at the recommended daily oral dose). Desloratadine had no effect on pup development at an oral dose of 3 mg/kg/day (estimated Desloratadine and Desloratadine metabolite exposures were approximately 7 times the AUC in humans at the recommended daily oral dose.
- Food And Drug Interaction
The plasma pharmacokinetics of desloratadine are weakly altered by coadministration of P-gp inhibitors (e.g. ketoconazole, azithromycin, erythromycin and itraconazole) or inducers (e.g. rifampicin [rifampin])(Molimard et al.,2004). Desloratadine is not a substrate of OATP (particularly OATP1A2). Grapefruit, apple and orange juice interfere with P-gp/OATP activity. (Dresser, et al.,2005).Studies comparing the effects of coadministering either desloratadine with grapefruit juice found the pharmacokinetics of desloratadine to be essentially unchanged. Coadministration of desloratadine with food or antacids does not affect its absorption.
In pharmacokinetic and clinical trials, desloratadine was coadministered with numerous CYP3A4 and CYP2D6 inhibitors (specifically, erythromycin, ketoconazole, azithromycin, cimetidine and fluoxetine), with no effects on clinically relevant parameters.(Aerius 2006) (Gupta , Banfield , Kantesaria et al., 2004). A number of drugs and other substances that are prone to interactions, such as ketoconazole, erythromycin and grapefruit juice, have shown no influence on desloratadine concentrations in the body. Desloratadine is judged to have a low potential for interactions.
- Use Of Desloratadine In Special Patient Subpopulations
The clinical efficacy and safety of desloratadine was evaluated in >2300 patients aged 12–75 years with SAR. A total of 1838 patients received 2.5–20 mg/day of desloratadine in four random- ized, double-blind, placebo-controlled clinical trials of 2–4 weeks’ duration conducted in the US.( 2005) The mean Cmax and AUC values for desloratadine following multiple-dose administration were 20% greater in younger subjects (aged <65 years) than in older subjects (aged 65 years). The apparent oral clearance, when normalized for bodyweight, was similar between the two age groups. The mean t1/2 of desloratadine was 33.7 hours in subjects aged 65 years. The pharmacokinetics for 3-hydroxydeslorata- dine (3-OH desloratadine) appeared to be unchanged in older versus younger subjects. Although age-related differences have been observed, they are not clinically relevant, and no dosage adjustment is required in elderly patients( 2005). Although the Cmax and AUC values with desloratadine and 3-OH desloratadine were higher in females than in males, no clinical relevance that requires dosage adjustment has been demonstrated.
Desloratadine pharmacokinetics following a single dose of 7.5 mg were characterized in subjects with mild (n = 7; creatinine clearance [CLCR] 51–69 mL/min/1.73 m2), moderate (n = 6; CLCR 34–43 mL/min/1.73 m2) and severe (n = 6; CLCR 5–29 mL/min/ 1.73 m2) renal impairment or haemodialysis-dependent subjects (n = 6). In subjects with mild or moderate renal impairment, the median Cmax and AUC values increased by approximately 1.2- and 1.9-fold, respectively, relative to subjects with normal renal function. In subjects with severe renal impairment, or in those who were haemodialysis dependent, the Cmax and AUC values of desloratadine increased by approximately 1.7- and 2.5-fold, respectively. Minimal changes in 3-OH desloratadine concentrations were observed. Desloratadine and 3-OH desloratadine were poorly removed by haemodialysis. Plasma protein binding of deslorata- dine and 3-OH desloratadine was unaltered by renal impairment. Although the US label for desloratadine recommends adjusting the dosage for patients with renal impairment, the EU label advises caution in the case of severe renal insufficiency.(2005)
Subjects with hepatic impairment (n=12), regardless of severity, who were administered desloratadine had approximately a 2.4-fold increase in the AUC as compared with subjects with normal hepatic function (2005). The apparent oral clearance of desloratadine in subjects with mild, moderate and severe hepatic impairment was 37%, 36% and 28% of that in normal subjects, respectively. An increase in the mean t1/2 of desloratadine in subjects with hepatic impairment was observed. For 3-OH desloratadine, the mean Cmax and AUC values for subjects with hepatic impairment were not statistically significantly different from those in subjects with normal hepatic function. In a phase I, open-label, multiple-dose study of desloratadine 5 mg, increases in the Cmax and AUC were observed in subjects with moderate hepatic impairment, consistent with findings from single-dose studies(2005). When factors such as race and metabolizer status were eliminated, the median exposure to desloratadine in subjects with hepatic impairment was about 3-fold greater than in subjects with normal liver function. On the US desloratadine label, dosage adjustment is recommended for patients with hepatic impairment; no adjustments are mentioned on the EU label(2006).
- Poor Metabolizers
Phenotypic polymorphism in the metabolism of desloratadine has been observed in those individuals with a decreased ability to form 3-OH desloratadine, the major metabolite of
desloratadine. Such individuals are termed ‘poor metabolizers of desloratadine. The prevalence of the poor metabolizer phenotype in the general population is approximately 6%; however, the prevalence among African Americans is 17% higher than that of any other population subgroup. The increased exposure in poor metabolizers is independent of age when administered at age- appropriate doses and is not associated with any changes in the safety and tolerability profile of desloratadine.(Prenner,Kim,Gupta, et al. 2006).
- Pharmaceutical Technology
- Dosage Form : Tablet , Syrup
- Available Dosage: Oral dosage
- Dosage: Tablet: 5mg, Syrup: 0.5mg
- Quality Parameters Tested
- Thickness and diameter
Tablet thickness and diameter should be controlled to ensure uniformity in tablets appearance and fitting into the containers for packaging process. If certain tablets are thick than the others, the given number of tablets are no longer the same as the volume of given bottle size. Tablets thickness and diameter also important in counting tablets using the filling equipment as it is use as counting mechanism.
- Hardness
Hardness is also so called crushing strength. It is the load required to crush the tablet when placed on its edge.
Tablets must be able to withstand the rigors of handling and transportation experienced in the manufacturing plant, in the drug distribution system, and in the field at the hands of the end users (patients/consumers). Manufacturing processes such as coating, packaging, and printing can involve considerable stresses, which the tablets must be able to withstand. For these reasons, the mechanical strength of tablets is of considerable importance and is routinely measured.
The principle of measurement involves subjecting the tablet to an increasing load until the tablet breaks or fractures. The load is applied along the radial axis of the tablet. Hardness is nothing but crushing strength. The resistance of tablets to capping, abrasion or breakage under conditions of
storage, transportation and handling before usage depends on its hardness. It is necessary because :
- Hardness can affect the disintegration. So if the tablet is too hard, it may not disintegrate in the required period of time.
- If the tablet is too soft, it will not withstand the handling during subsequent processing such as coating and packaging (Banker and Anderson, 1986).
- Friability Test
Friability refers the ability of the compressed tablet to avoid fracture and breaking during transport. Friability is defined as the percent of weight loss by tablets due to mechanical action during the test. Tablets are weighing before and after testing and friability is expressed as a percentage loss on tablet weight.
The tablets are well subjected to a uniform tumbling motion to check its strength, proper binding of powder, lack of elegance and ability of tablet to withstand mechanical shock of tablet handling in manufacturing, packing and shipping. coating, packaging, transport, which are not severe enough to break the tablet, but may abrade the small particle from tablet surface. To examine this, tablets are subjected to a uniform tumbling motion for specified time and weight loss is measured. An instrument called friabilator is used to evaluate the ability of the tablet to withstand abrasion in packaging, handling, and shipping.
Friability is defined as the percent of weight loss by tablets due to mechanical action during the test. A tablet is weighing before and after testing and friability is expressed as a percentage loss on pretest tablet weight.
- Weight Variation
Weight variation test is performed to check that the manufactured tablets uniformity. This test is performed by SHIMADZU Electric Balance.
To evaluate tablets potential for efficacy, the amount of drug per tablet needs to be monitored. A tablet contains a specific amount of drug in a specific amount of tablet weight. So tablet weight
and its uniformity may help to ensure that a tablet contains the proper amount of drug. For these weight variation test is performed.
Weight variation test is performed to check whether the manufactured tablets have an uniform weight.
13 tablets were weighed individually and a compendia weight was taken. The average weight was obtained by dividing the compendia weight by 13, then the average weight was compared to the individual weight.
- Disintegration test
Disintegration is the time required for the tablet to break into particles, the disintegration test is a measure only of the time required under a given set of conditions for a group of tablets to disintegrate into particles.
The disintegration test is performed to find out the time it takes for a solid oral dosage form like a tablet or capsule to completely disintegrate. The time of disintegration is the measure of the quality. This is because, for example, if the disintegration time is too high, it means that the tablet is highly compressed. If the disintegration time is not uniform in asset of sample being analyzed, it indicates batch inconsistency and lack of batch uniformity (Banker and Anderson, 1986).
- Dissolution
Dissolution may be defined as the amount of drug substance that goes into solution under standardized conditions of liquid/solid interface, temperature and solvent composition.
Active absorption of oral dosage forms depends on adequate release of the active pharmaceutical ingredient (API) from the product. Dissolution or solubility of the API play pivotal role in this aspect. Dissolution testing is used as a tool to identify a crucial effect in the bioavailability of the API.In biological system, drug dissolution in an aqueous medium is an important polar condition of systemic absorption. The rate at which drugs with poor solubility dissolve from an intact or disintegrated solid dosage form in the gastrointestinal tract often controls the rate of the systemic absorption of the drug. Thus dissolution tests are discriminating factors that may affect drug bioavailability (Aldeborn, 1988).
- purpose of the study
Substandard drugs product can be defined as genuine drugs manufactured by authorized manufactures but do not meet the quality specification fixed for them by national standard (world Health Organization, 2013). Thus, monitoring of generic drugs in the market in vital. WHO has issued many guidelines for the assessment, authorization , registration, marketing as well as quality assurance of the generic drugs products (Michael et al.,2003). Monitoring marketed drugs can lessens a country’s economical problem on health issue from disease due to fraud and substandard drugs usage ( Musa et al.,2011).
The aim of this study was to analyze and compare three different brands of 5mg desloratadine tablets, commercially available in the local market of Bangladesh, by evaluating the tablets diameter, thickness, hardness, friability, disintegration, dissolution and potency in order to assume their pharmaceutical quality.
Chapter 2
Materials and Methods
- Materials and Methods
Collection of Sample: Commercially available six different brands of desloratadine 5mg film coated tablets were purchased from the retail pharmacies, Lazz Pharma and Shohag Pharma, in Dhaka city. 20 tablets of each brand were purchased after the samples were properly checked for their physical appearance, name of manufacturer, manufacturing date, expiry date, manufacturing license number and batch number at the time of purchase.
The brands were given identifiable codes as A, B,and C, and the study was carried out at the Biopharmaceutics Laboratory of Pharmacy department of University of Asia Pacific.
Collection of Standard (API): Standard sample of desloratadine powder were collected from ACI Pharmaceuticals Limited, Bangladesh.
Collection of other Chemicals: All other chemicals used were available from the Biopharmaceutics Laboratory of Pharmacy department of University of Asia Pacific.
- Apparatus
Table 2 : Apparatus used for the test
| In vitro Test | Main Apparatus | Manufacturer |
| Weight variation | Analytical balance | AY-200, Shimadzu, Japan. |
| Diameter and Thickness | Digital Slide calipers | Shanghai, China. |
| Friability | Roche friabilator | VFT-2, Veego, India. |
| Hardness | Digital tablet Hardness tester | 8M, Dr Schleuniger, Switzerland. |
| Disintegration | Tablet disintegration tester (USP device) | VDT-2, Veego, India. |
| Dissolution | USP dissolution apparatus II (Paddle apparatus) | TDT-08L, Electrolab, SAKA International Ltd. |
| Potency | UV vis spectrophotometer | UVmini-1700, Shimadzu, Japan. |
Other apparatus used include-
- Filter paper
- 100 ml volumetric flask
- 500 ml volumetric flask
- 1000 ml measuring cylinder
- 500 ml beaker
- 1000 ml beaker
- Conical flasks
- Test tubes
- Test tube racks
- Pipettes (1ml, 5ml, and 10ml)
- Pipette filler
- Spatula
- Mortar and pestle
- Filter funnel
- Thermometer
- Glass rod
- Wax paper
- Weight Variation (Uniformity of Weight) Test
Procedure: 20 desloratadine tablets from each of the 6 brands were selected at random and using an analytical balance each one was weighed individually, W1, W2, W3… W20. The average weight was determined using the formula, Wavg = (W1+W2+W3+…+W20)/20.
The percent deviation from average weight may be calculated by following way:
Weight Variation = Individual weight — Average weight X 100
Average weight

Figure 2.1: Electronic Balance
- Thickness Test
Digital Slide Caliper: It consists of external jaws between which the tablet is placed and contains a locking screw to move the jaws. An LCD monitor displays the reading in mm or inch as chosen. It also contains the power button, zero setting and inch/mm button.
Procedure: 20 desloratadine tablets of each brand were individually placed between the 2 jaws of a digital slide caliper. These jaws were then closely fitted to tablet along its height and the locking screw was tightened so that the jaws do not move apart. The display reading was recorded.
The average thickness was determined for each brand using the formula:
Average Thickness = Sum of total thickness of all tablets / No. of tablets The percent of thickness variation was then calculated by the following way:
Thickness Variation = Individual thickness—Average thickness X 100
Average thickness

Figure 2.2: Digital Slide Calipers
- Diameter Test
Procedure: 20 desloratadine tablets of each brand were placed between the 2 jaws of a digital slide caliper and then these jaws were closely fitted to tablet along its diameter and the reading was recorded.
The average diameter was determined for each brand using the formula:
Average Diameter = Sum of total diameter of all tablets / No. of tablets The percent of diameter variation was then calculated by following way:
Diameter Variation = Individual diameter — Average diameter X 100
Average diameter
- Friability Test
Roche Friabilator: The Friabilator consists of a plastic chamber divided into two parts which revolves at 25 rpm, dropping the tablets a distance of 6 inches with each revolution.
Procedure: 7 tablets of each brand were weighed together = W1. These tablets were subjected to abrasion by employing a Veego Friabilator (VFT-2, India) operated for 100 revolutions at 25 rpm for 4 minutes. After dedusting the tablets, only the intact ones were weighed together = W2.
% Friability was calculated by using the following formula,
% Friability (% loss) = {(W1 – W2) / W1} x 100

Figure 2.3: Electrolab Friabilator
- Hardness Test
Procedure: A tablet was placed between the two pressure plates. One of the plates moves in order to damage the tablet. Measurement is carried out on 3 tablets, taking care to remove all the fragments of the broken tablets before each determination.
The average hardness is then calculated using the formula,
Average Hardness = Sum of hardness of all tablets / Total number of tablets

Figure 2.4: Hardness Tester
7. Disintegration Test
Preparation of Disintegration Medium (0.1N HCl): Our disintegration medium was 0.1N HCl solution having a pH of 1.2. As we know, 34.5 gm HCl is present in 1300 ml solution having 1 normality, thus in 1300 ml 0.1N HCl solution, 3.45gm will be present. We needed to prepare 24 liters of medium for the disintegration test. Because 37% HCl was available in our laboratory and we know that 9.86 ml of 37% HCl is present in 1L solution, we dissolved 234.64 ml of 37% HCl in 22 liters of distilled water and adjusted the volume up to 24 liters to prepare the medium of our desired volume. The pH was then checked and adjusted to 1.2.
8. Disintegration Apparatus: The U.S.P. disintegration apparatus employs 6 transparent glass tubes, 6 inches long, and which are open at the top and held against a 10 mesh screen at the bottom end of the basket rack assembly. In practice, 1 tablet is placed in each tube and the basket rack is positioned in a 1 liter beaker containing disintegration medium. A standard motor driven basket is used to move the basket rack assembly up and down through a distance of 5-6 cm at a frequency of 28-32 cycles per minute. The tablets should remain at least 2.5 cm below the liquid surface on their upward movement and descend to not closer than 2.5 cm from the bottom of beaker. Perforated plastic discs are placed on top of the tablets to prevent them from floating out of tubes (Banker, 1974).
Procedure: Disintegration test was carried out on 3 tablets of each brand using a tablet disintegration apparatus (Veego, India). 600 ml of 0.1N HCl medium was placed in the beaker of the apparatus. 3 tablets of each brand were placed in 3 tubes out of the 6 tubes of the basket with one tube between two empty and the basket was immersed in 0.1N HCl medium. The apparatus was then operated at 37 ± 2° C and the time required for each tablets to disintegrate were recorded. Average disintegration time for each brand was calculated using the formula:
Average Disintegration Time = Sum of disintegration times of all tablets of a brand
No. of tablets in each brand
- Dissolution Test
Preparation of Dissolution Medium (0.1N HCl): Since our dissolution and disintegration media are the same, 34 liters of 0.1N HCl for dissolution was prepared in the same way as was employed for preparing the disintegration medium.
Procedure: At first 900 ml 0.1N HCl solution was transferred into 3 vessels of dissolution machine. Besides 1300 ml 0.1N HCl solution was kept as a reservoir. Temperature was set at 37ºC using USF type II apparatus(paddle) and the rotation of paddle was set at 50 rpm samples withdraw at 5,10,15,30 and 60 . The paddle was fitted to the machine and 3 tablets of a brand were then introduced individually in the vessels of the system. The machine along with a stopwatch was turned on simultaneously. After every 10 minutes interval for 1 hour, 10 ml solution was drawn out from each vessel which was again recovered with the 0.1N HCl solution. The collected solution was then filtered so that we get the active other than additives. This solution was next run through UV visible spectrophotometer and the absorbance (y) was taken at λmax 281 nm. To determine the concentration (x) of sample, the equation obtained from the standard curve (y = 0.034x + 0.025) of pure API was used. This process was repeated for 3 tablets from each of the 5 remaining brands.
Drug content can be calculated from the following formula:
Drug Content =
Absorbancesample X Dilutionstandard X Potency of standard X Average weight Absorbancestandard X Dilutionsample
% release of drug can then be calculated using the following formula:
% Release of Drug = (Drug content / Therapeutic value) X 100
- Potency Test
- Preparation of Standard Curve
Preparation of Stock Solution: 20 mg of standard desloratadine powder (API) was taken in a 100 ml volumetric flask and dissolved in 0.1N HCl up to 100 ml. 10 ml of this solution was taken in another 100 ml volumetric flask and diluted up to 100 ml with 0.1N HCl. This is the stock solution.
Preparation of Standard Solution for Standard Curve: 10 test tubes were taken and marked serially from 1-10 and then kept in a test tube rack. In test tube marked 1, 1 ml of this stock solution and 9 ml 0.1N HCl was added; in test tube 2, 2 ml stock solution and 8 ml 0.1N HCl was added. This was continued by serially moving off to test tube 10 with 1 ml increase in stock solution and 1 ml decease in 0.1N HCl in each of the subsequent test tubes to keep a constant volume of 10 ml in all tubes. Hence, test tube 10 contained only 10 ml stock solution only (concentration= 20 mcg/ml). Thus the standard solutions were prepared.
- Determination of Absorbance: The cell was filled with the standard solution from test tube 1 and then placed into the sample holder of UV vis spectrophotometer and the absorbance was noted at 281 nm. In the same way, absorbances of the other working standards in the remaining 9 tubes were determined.
- Maximum wave length absorption determination : Standard solution of 0.1 mg/ml (100µg/ml) desloratadine were prepared in 0.1 M HCl (PH =1.2). The sample was submitted to UV spectrophotometer between (400-200nm) to determine the maximum. Wave length absorption (ʎmax=281) of determine.
- Determination of Standard Curve: A graph of absorbance against concentration is plotted in Microsoft Excel which also represents the R2 value and equation of the graph.
Table 3 : Concentration of drugs for standard curve
| Test Tube Number | Stock Solution (ml) | Concentration (µg/ml) | Absorbance |
| 1 | 1 | 3.4µg | 0.147 |
| 2 | 2 | 6.8µg | 0.207 |
| 3 | 3 | 10.2µg | 0.265 |
| 4 | 4 | 13.6µg | 0.380 |
| 5 | 5 | 17µg | 0.457 |
| 6 | 6 | 20.4µg | 0.543 |
| 7 | 7 | 23.8µg | 0.691 |
| 8 | 8 | 27.2µg | 0.718 |
| 9 | 9 | 30.6µg | 0.809 |
| 10 | 10 | 34µ | 0.885 |
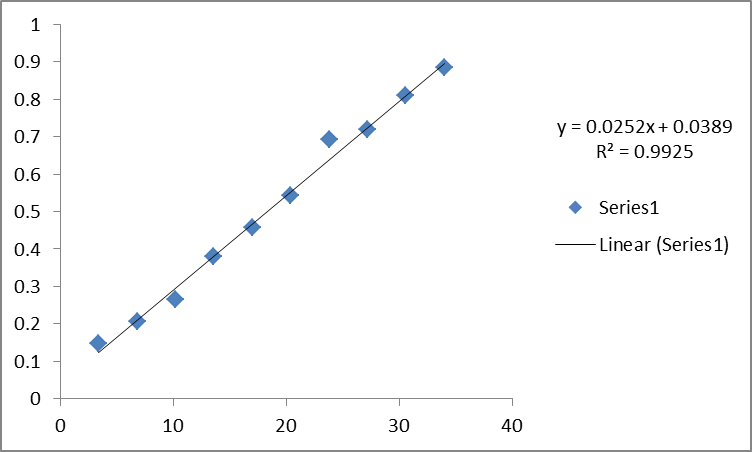
Figure 2.1: Standard curve of desloratadine
- Preparation of Sample Solution: 4 tablets of a brand are weighed individually and their average weight (equivalent to 5 mg) is calculated. The tablets are crushed in a mortar with pestle and 20 mg equivalent weight of powder is measured. This is taken in a 100 ml volumetric flask and dissolved in small amount of 0.1N HCl and the volume is made up to 100 ml. 1 ml of this solution is taken in a test tube and diluted with 9 ml 0.1N HCl. This is the sample solution (concentration= 20 mcg/ml). The process is repeated for the remaining 5 brands.
- Determination of Absorbance: Two-third of the cell was filled with the sample solution and then placed into the sample holder of UV spectrophotometer and the absorbance was noted at 281 nm.
- Calculation of Potency: The drug content can be calculated by using the formula, Drug Content =
Absorbancesample X Dilutionstandard X Potency of standard X Average weight Absorbancestandard X Dilutionsample
Percent potency can then be calculated by using the formula,
% Potency = (Drug content / Therapeutic value) X 100
Chapter -3
Results and Discussion
- Aesthetic test
Since the study was conducted with tablets from six different brands it possess different aesthetic appearance. The results of aesthetic assessment of the tablets have been given below.
Table 4 : Results of aesthetic tests of the tablets of three different brands
| Sample | Color | Shape | Lustre | Nature of Surface |
| A | Dark blue | Crystal shape Convex Bisect | Dull | Smooth |
| B | Pinkish | Round shape | Dull | Smooth |
| C | Orange | Round shape Convex Bisect not Flush | Dull | Smooth |
- Friability
Result: The following graph represents the % friability of 3 brands.
Figure: Comparison of tablet friability of three different brands
| Brand name | % friability |
| A | 1.63 |
| B | 0.262 |
| C | 0.145 |
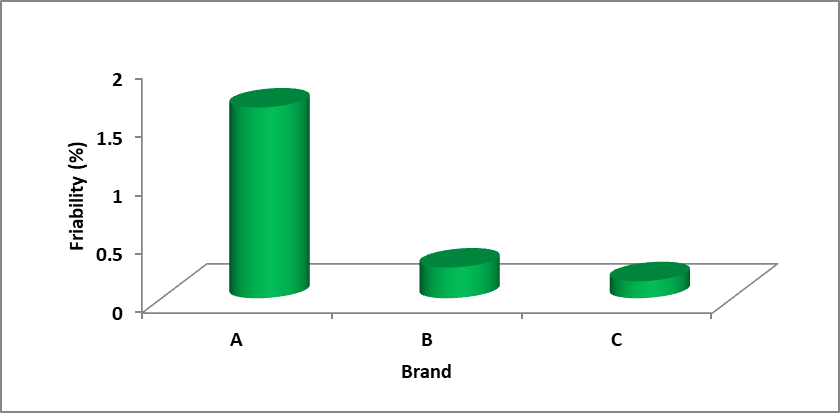
Figure: Comparison of tablet friability of three different brands
Discussion: Conventional compressed tablets that loos less than 0.5 – 1.0% of their weight are generally considered acceptable (Lachman et al.,1999). Here all the tablets met the specifications and all the brands having percent friability below 0.5% . Among all the brands, brand-C had the lowest percentage of friability and it was 0.14% and brand-A had the highest percentage of friability and it was 1.63%.
- Thickness
Result: The average thickness of 3 brands and their positive and negative deviation of thickness are given bellow.
Table 5 : Comparison of the thickness of 3 different brands of desloratadine.
| Brand name | Avg Thickness (mg) | Max deviation | Min deviation |
| A | 3.080295858 | 0.8984 | -1.372 |
| B | 3.127218935 | 18.594 | 1.1049 |
| C | 3.700236686 | 3.0801 | -0.246 |
Discussion: Tablet thickness should be controlled within a ± 5 % variation of a standard value (Lachman et al.,1999).Considering the average thickness as standard, all the tablets were within
the variation range of ± 5 %. The brand – B showed highest maximum and minimum deviation from average value.
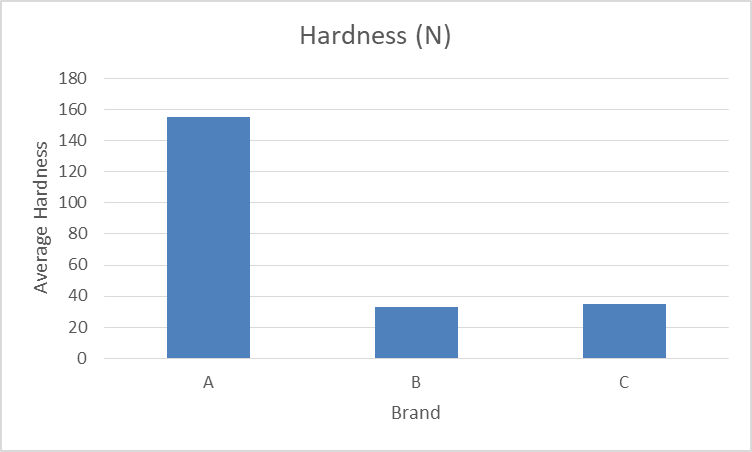
- Hardness
Hardness of tablet depends on the amount of binder used in the granulation process. It also depends on the pressure applied during the process of compression.
| Brandname | Hardness(N) |
| A | 155 |
| B | 33 |
| C | 35 |
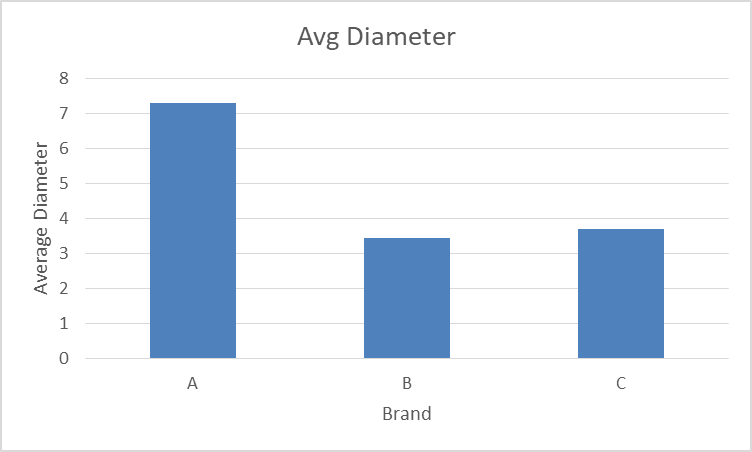
- Diameter
The following value represents the comparative study of diameter of desloratadine (tablet) of three different marketed brands in which 20 tablets of each brand were taken.
| Brand name | Avg Diameter |
| A | 7.28816568 |
| B | 3.445739645 |
| C | 3.700236686 |
- Potency
Result: A comparative representation of potency of 3 different brands has been given by the following graph.
| Brand name | potency (%) |
| A | 74.13 |
| B | 102.87 |
| C | 126.82 |
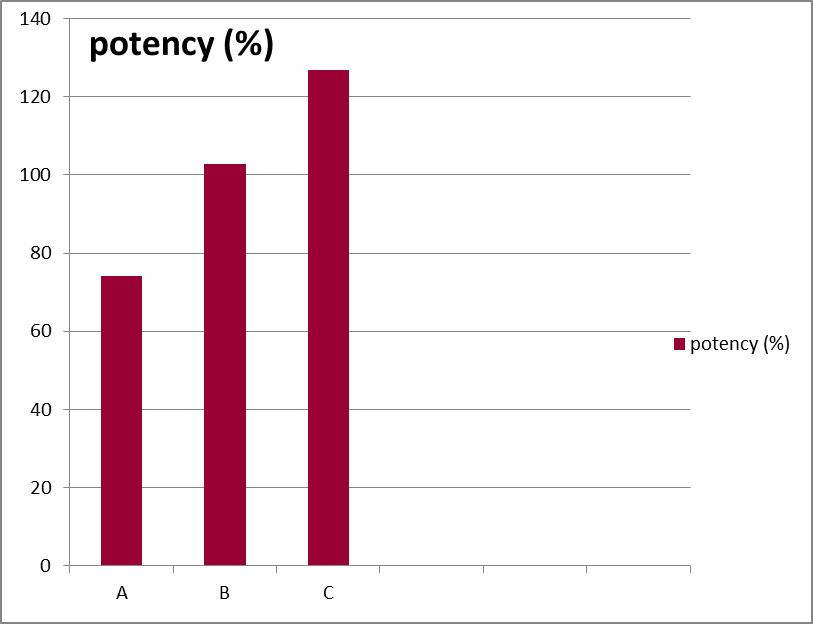
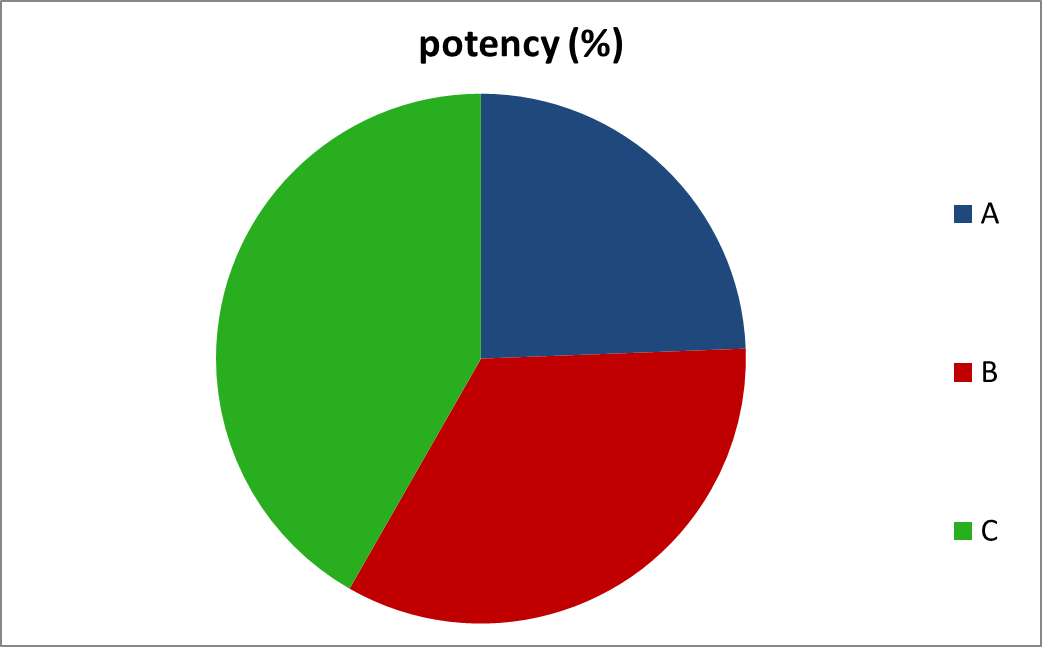
Figure: Comparison of the potency of 3 different brands of desloratadine.
Discussion: Official compendia or other standards provide an acceptable potency range around the libil potency. For highly potent, low dose drug this range is usually not less than 90% and not more than 110% of the labeled amount. For most other larger dose drugs in tablet form. The official potency range that is permitted is not less than 95% and not more than 105% of labeled amount (Lachman et al.,1999).
Our study was conducted with low dose desloratadine (5 mg), so the percent potency should be within( 90 -110) %. It is observed that all brands met this specification except brand–A possessed 74.13% potency.
- Conclusion
We performed some simple in vitro tests in order to compare the multi brand generic molecules for having good therapeutic activity. Unlike band A and B, Band C did not meet the USP weight variation specification and have average diameter of 13.27 mm. Average length and width of brand A is 14.17 mm and 7.16 mm respectively whereas average diameter of brand B is 11.63 mm. These variations in diameter reveal that different brands vary in shape and size to have different aesthetic appeal. Average thickness of all brands are between 3.80-4.10 mm.
Reference
Anthes, J.C., Gilchrest, H., Richard, C., Eckel, S., Hesk, D., West Jr, R.E., Williams, S.M., Greenfeder, S., Billah, M., Kreutner, W. and Egan, R.W., 2002. Biochemical characterization of desloratadine, a potent antagonist of the human histamine H1 receptor. European journal of pharmacology, 449(3), pp.229-237.
Kreutner, W., Hey, J.A., Anthes, J., Barnett, A., Young, S. and Tozzi, S., 2000. Preclinical pharmacology of desloratadine, a selective and nonsedating histamine H1 receptor antagonist. Arzneimittelforschung, 50(04), pp.345-352.
Gillard, M., Van Der Perren, C., Moguilevsky, N., Massingham, R. and Chatelain, P., 2002. Binding characteristics of cetirizine and levocetirizine to human H1 histamine receptors: Contribution of Lys191and Thr194. Molecular pharmacology, 61(2), pp.391-399.
Bakker, R.A., Wieland, K., Timmerman, H. and Leurs, R., 2000. Constitutive activity of the histamine H1 receptor reveals inverse agonism of histamine H1 receptor antagonists. European journal of pharmacology, 387(1), pp.R5-R7.
Wu, R.L., Anthes, J.C., Kreutner, W., Harris, A.G. and West Jr, R.E., 2004. Desloratadine inhibits constitutive and histamine-stimulated nuclear factor-κB activity consistent with inverse agonism at the histamine H1 receptor. International archives of allergy and immunology, 135(4), pp.313-318.
Wu, R.L., Anthes, J.C., Kreutner, W., Harris, A.G. and West Jr, R.E., 2004. Desloratadine inhibits constitutive and histamine-stimulated nuclear factor-κB activity consistent with inverse agonism at the histamine H1 receptor. International archives of allergy and immunology, 135(4), pp.313-318.
de Esch IJP, Thurmond RL, Jongjean A, . The histamine H4 receptor as a new therapeutic target for inflammation. Trends in Pharmacological Sciences 2005; 26: 462-469.
Liu, H. and Farley, J.M., 2005. Effects of first and second generation antihistamines on muscarinic induced mucus gland cell ion transport. BMC pharmacology, 5(1), pp.1-10.
Cardelús, I., Antón, F., Beleta, J. and Palacios, J.M., 1999. Anticholinergic effects of desloratadine, the major metabolite of loratadine, in rabbit and guinea-pig iris smooth muscle. European journal of pharmacology, 374(2),pp.249-254.
Henz, B.M., 2001. The pharmacologic profile of desloratadine: a review. Allergy, 56, pp.7-13.
Anolik, R., 2009. Desloratadine and pseudoephedrine combination therapy as a comprehensive treatment for allergic rhinitis and nasal congestion. Expert Opinion on Drug Metabolism & Toxicology, 5(6), pp.683-694.
Schroeder, J.T., Schleimer, R.P., Lichtenstein, L.M. and Kreutner, W., 2001. Inhibition of cytokine generation and mediator release by human basophils treated with desloratadine. Clinical & Experimental Allergy, 31(9), pp.1369-1377.
Genovese, A., Patella, V., De Crescenzo, G., De Paulis, A., Spadaro, G. and Marone, G., 1997. Loratadine and desethoxylcarbonyl‐loratadine inhibit the immunological release of mediators from human FcɛRI+ cells. Clinical & Experimental Allergy, 27(5), pp.559-567.
Agrawal, D.K., 2004. Anti‐inflammatory properties of desloratadine. Clinical & Experimental Allergy, 34(9), pp.1342-1348.
Telfast 120 and 180 [prescribing information ;online].Guildford: Sanofi-Aventis,2007Feb 20.AvailablefromURL:http://emc.medicines.org.uk/emc/assets/c/html/displaydoc.asp?.document id=6659 [Accessed 2008 Feb 18].
Cyr, M.M., Hayes, L.M., Crawford, L., Baatjes, A.J., Keith, P.K. and Denburg, J.A., 2005. The effect of desloratadine on eosinophil/basophil progenitors and other inflammatory markers in seasonal allergic rhinitis: a placebo-controlled randomized study. International archives of allergy and immunology, 138(3), pp.209-216.
Lippert, U., Möller, A., Welker, P., Artuc, M. and Henz, B.M., 2000. Inhibition of cytokine secretion from human leukemic mast cells and basophils by H1‐and H2‐receptor antagonists. Experimental dermatology, 9(2), pp.118-124.
Affrime, M., Gupta, S., Banfield, C. and Cohen, A., 2002. A pharmacokinetic profile of desloratadine in healthy adults, including elderly. Clinical pharmacokinetics, 41(1), pp.13-19.
Devillier, P., Roche, N. and Faisy, C., 2008. Clinical pharmacokinetics and pharmacodynamics of desloratadine, fexofenadine and levocetirizine. Clinical pharmacokinetics, 47(4), pp.217-230.
Xyzal[prescribing information; online].Slough:UCB Pharma Limited,2007Jul.Available from URL:http://emc.medicines.org.uk/industry/default.asp?.page=displaydoc.asp&documentid=1987 7[Accessed 2008 Feb 18].
Devillier, P., Roche, N. and Faisy, C., 2008. Clinical pharmacokinetics and pharmacodynamics of desloratadine, fexofenadine and levocetirizine. Clinical pharmacokinetics, 47(4), pp.217-230.
Day, J.H., Briscoe, M., Rafeiro, E., Chapman, D. and Kramer, B., 2001. Comparative onset of action and symptom relief with cetirizine, loratadine, or placebo in an environmental exposure unit in subjects with seasonal allergic rhinitis: confirmation of a test system. Annals of Allergy, Asthma & Immunology, 87(6), pp.474-481.
Horak, F., Jäcer, S. and Berger, U., 1992. Onset and duration of the effects of three antihistamines in current use–astemizole, loratadine and terfenadine forte–studied during prolonged, controlled allergen challenges in volunteers. Journal of international medical research, 20(5), pp.422-434.
Molimard, M., Diquet, B. and Benedetti, M.S., 2004. Comparison of pharmacokinetics and metabolism of desloratadine, fexofenadine, levocetirizine and mizolastine in humans. Fundamental & clinical pharmacology, 18(4), pp.399-411.
Barecki, M.E., Casciano, C.N., Johnson, W.W. and Clement, R.P., 2001. In vitro characterization of the inhibition profile of loratadine, desloratadine, and 3-OH-desloratadine for five human cytochrome P-450 enzymes. Drug metabolism and disposition, 29(9), pp.1173-1175.
Banfield, C., Herron, J., Keung, A., Padhi, D. and Affrime, M., 2002. Desloratadine has no clinically relevant electrocardiographic or pharmacodynamic interactions with ketoconazole. Clinical pharmacokinetics, 41(1), pp.37-44.
Affrime, M., Banfield, C., Gupta, S., Cohen, A., Boutros, T., Thonoor, M. and Cayen, M., 2002. Effect of race and sex on single and multiple dose pharmacokinetics of desloratadine. Clinical pharmacokinetics, 41(1), pp.21-28.
Gupta, S., Banfield, C., Kantesaria, B., Flannery, B. and Herron, J., 2004. Pharmacokinetics/pharmacodynamics of desloratadine and fluoxetine in healthy volunteers. The Journal of ClinicalPharmacology, 44(11),pp.1252-1259.
Liu, G.Z., Xu, H.W., Chen, G.W., Wang, P., Wang, Y.N., Liu, H.M. and Yu, D.Q., 2010. Stereoselective synthesis of desloratadine derivatives as antagonist of histamine. Bioorganic & medicinal chemistry, 18(4), pp.1626-1632.
Fischer, J. and Ganellin, C.R., 2010. Analogue-based drug discovery. Chemistry International–Newsmagazine for IUPAC, 32(4), pp.12-15.
See, S., 2003. Desloratadine for allergic rhinitis. American Family Physician, 68(10), p.2015.
Blaess, M., Kaiser, L., Sommerfeld, O., Csuk, R. and Deigner, H.P., 2021. Drugs, metabolites, and lung accumulating small lysosomotropic molecules: Multiple targeting impedes SARS-CoV-2 infection and progress to COVID-19. International Journal of Molecular Sciences, 22(4), p.1797.
Lee, H.E., Chang, I.K., Lee, Y., Kim, C.D., Seo, Y.J., Lee, J.H. and Im, M., 2014. Effect of antihistamine as an adjuvant treatment of isotretinoin in acne: a randomized, controlled comparative study. Journal of the European Academy of Dermatology and Venereology, 28(12),
Share this content:
Přijetí hypoteční platby může být nebezpečný pokud nemáte rádi čekání
v dlouhých řadách , vyplnění mimořádné formuláře , a odmítnutí úvěru na základě vašeho úvěrového skóre .
Přijímání hypoteční platby může být
problematické, pokud nemáte rádi čekání v dlouhých řadách , podávání extrémních formulářů , a odmítnutí úvěru na základě vašeho úvěrového skóre .
Přijímání hypoteční platby může být problematické , pokud nemáte
rádi čekání v dlouhých řadách , vyplnění extrémních formulářů a odmítnutí úvěrových rozhodnutí založených na úvěrových skóre
. Nyní můžete svou hypotéku zaplatit rychle a efektivně
v České republice. https://groups.google.com/g/sheasjkdcdjksaksda/c/vq7BSyzEHsM
Thank you for your valuable comment.
Thank you for your comment. please support me.